Hey guys! Today we’re going to go over treatments for atrial fibrillation.
Suggested pre-readings so we’re all on the same page:
If you are interested in more anticoag stuff regarding afib, see the following posts:
Atrial Fibrillation: Chronic Anticoagulation 💊
Atrial Fibrillation: Acute Anticoagulation Part ✌️
Rate versus Rhythm Control
When we’re talking about treating afib, there are two main strategies we can employ: rate control or rhythm control.
I feel like I learned this in school, but I low-key didn’t really understand what they actually meant. So, in order to get a good understanding of what that actually means, let’s boil it down.

Rate Control
Rate control does nothing to the afib itself.
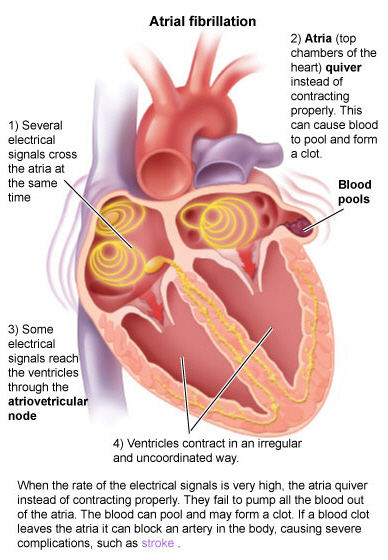
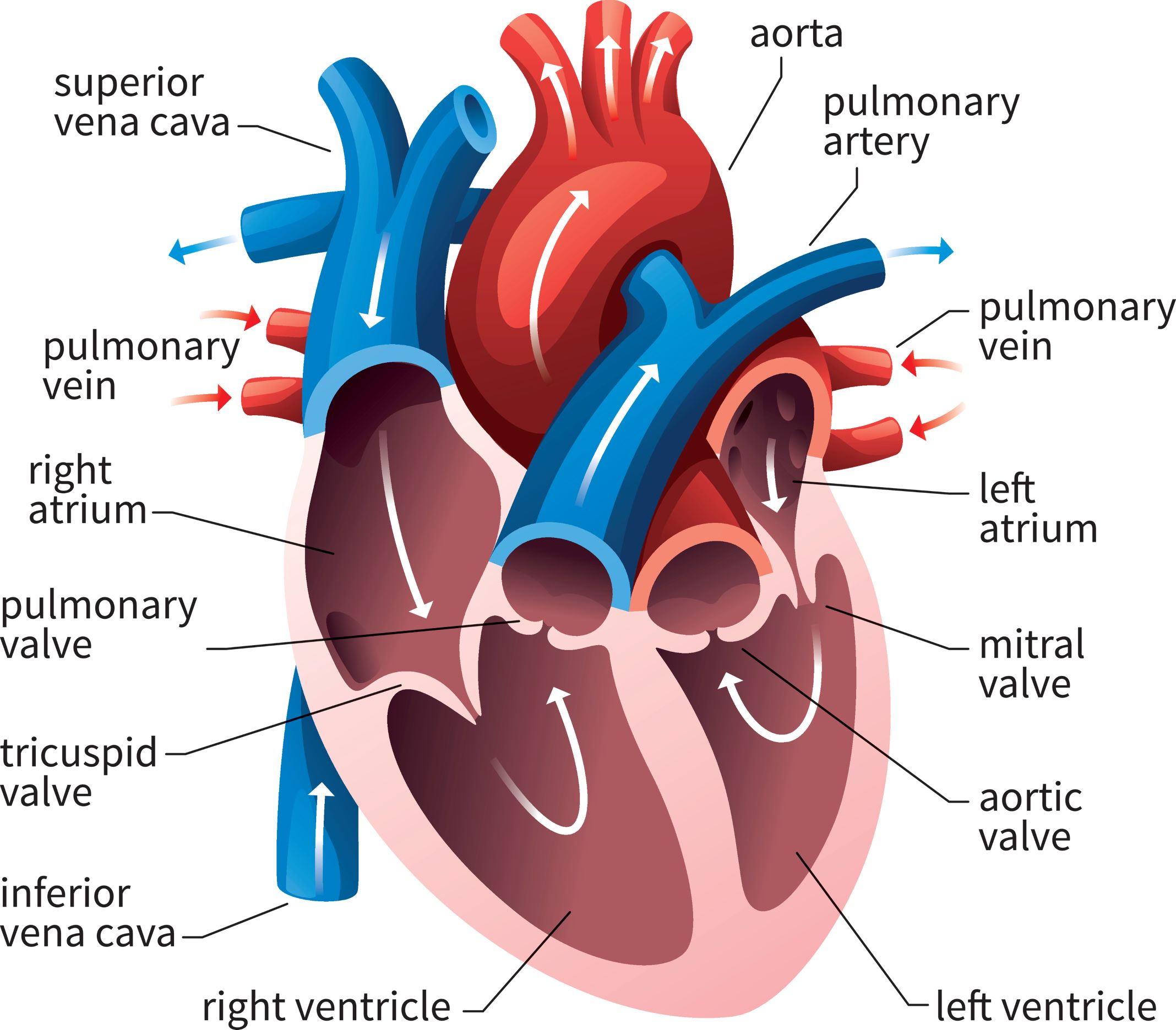
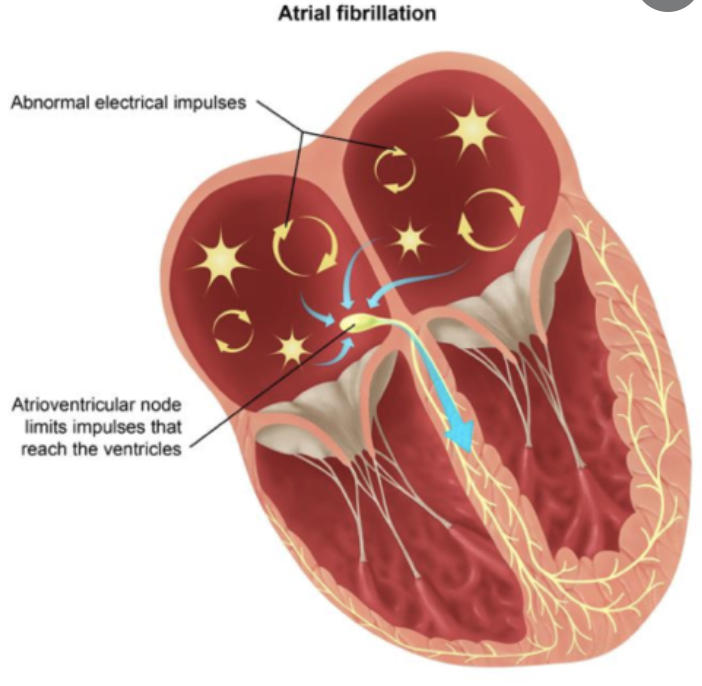
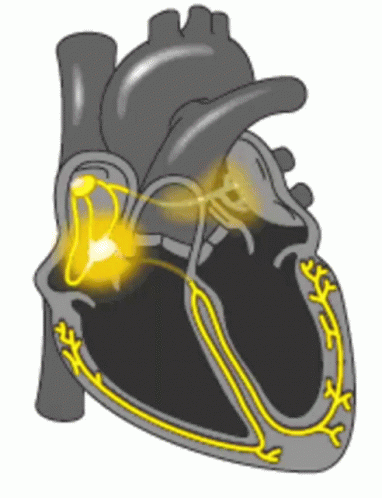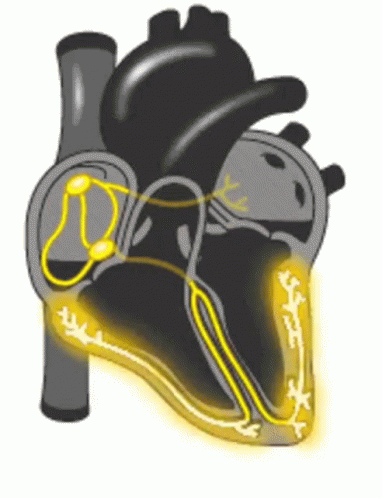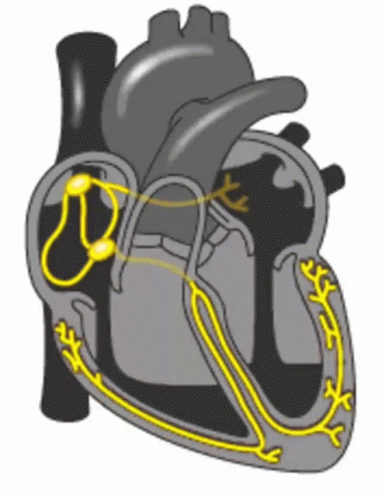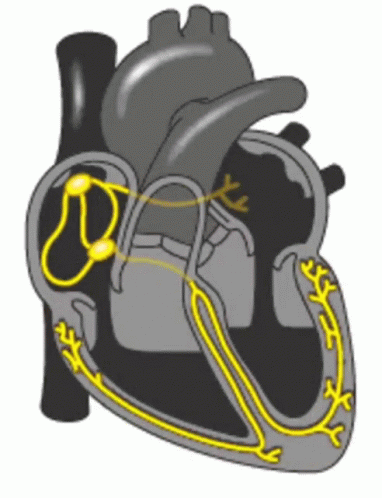
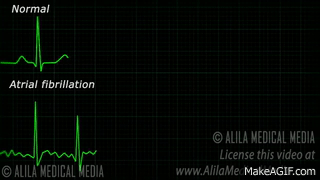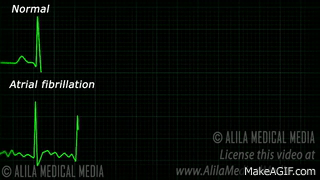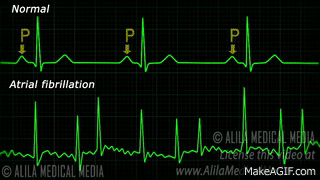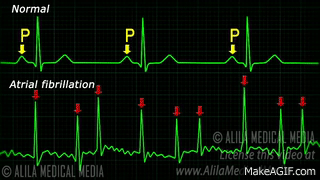
When a patient is “rate controlled”, if you looked into their heart, you’d still see those atypical ectopic points shooting off in their atria – in other words, the patient’s rhythm would still be afib – with undefined p waves on their EKG.

The goal of rate control is to control the ventricular rate.
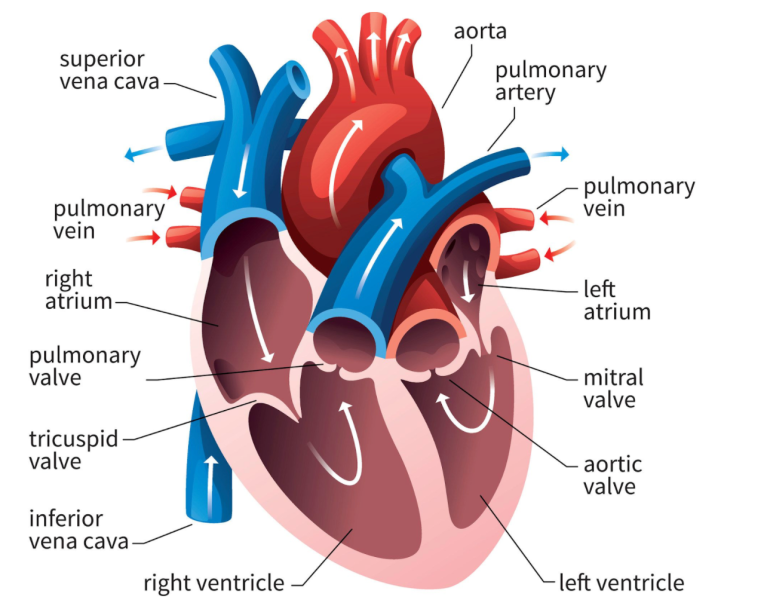
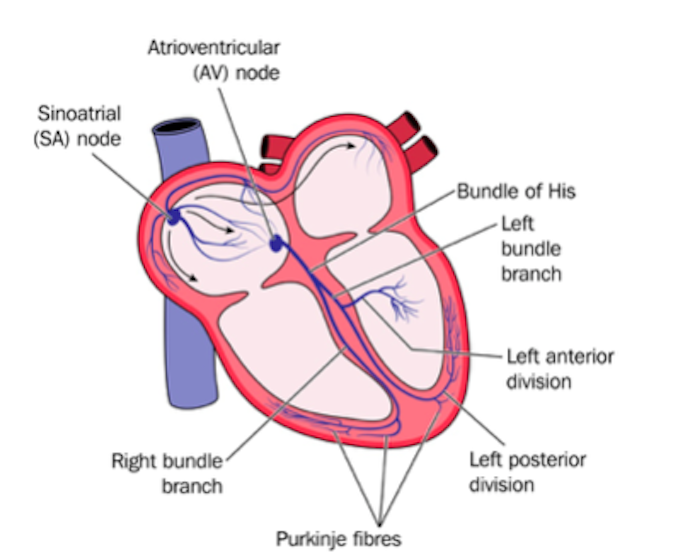
Remember how in our previous talk we said how some of those ectopic signals pass through that AV node and then cause the ventricles to contract?
And that, in the long term, that fast ventricular rate can cause issues like structural changes, remodeling, and even heart failure? Not to mention symptoms like dizziness.
By rate controlling an afib patient, we are making sure to keep that ventricular rate/systole under control. This will prevent dizziness, structural heart changes and remodeling.

Source: GIPHY
Rhythm Control
What about rhythm control?
In rhythm control, we’re actually attempting to convert the patient back into a normal sinus rhythm. This could be using a combo of strategies, including electrical cardioversion, using antiarrhythmic agents, or getting those areas of ectopy ablated in the atria.
All about Rate Control
Like we said above, rate control is when we give agents to try to decrease the conduction through that AV node and stop the ventricle from contracting so rapidly.
By doing this, we are helping patients avoid symptoms and also prevent them from getting a tachycardia-induced cardiomyopathy. Afterall, remember that your heart is a muscle. If I went to the gym (which I don’t) and start picking up weights and doing a bunch of reps what would happen to my arm muscles? They would grow right? Your heart is the same way. If faced with non-physiologic (aka not normal) conditions, it can undergo severe structural changes over time which can lead to heart failure.

Source: GIFTENOR
What is our target heart rate?
In rate control, we generally like to target a resting heart rate goal of < 80 beats per minute.
However, in patients that can’t tolerate such a low goal – and get dizzy – we may opt for a more lenient control strategy of < 110 bpm as long as these patients remain asymptomatic and their left ventricular systolic function is preserved .
Ok now we know what rate control is and why we do it.
Now let’s talk medicine.

In the realm of rate control we have the following potential agents in our arsenal:
- Beta blockers
- Non-dihydropyridine calcium channel blockers
- Digoxin
- Amiodarone
- Dronedarone
Hm, that seems like a lot right? How do I know which one to choose?

Ok don’t worry. Let’s first let’s start with our two first-line agents: beta blockers and non DHP calcium channel blockers.
You will also find that the decision to use one agent over another often depends on your patient and their comorbidities. Do they have heart failure? Do they have pulmonary issues like COPD? Are their conduction pathways normal?
Let’s get into it:
Beta Blockers
Beta blockers work to primarily block the beta-1 receptors located in the heart. Normally, our endogenous catecholamines epinephrine and norepinephrine can bind to these beta 1 receptors and cause an increase in heart rate as part of our “fight or flight” response.

These agents are available both orally and intravenously so can be used in both chronic (long term) and acute atrial fibrillation.
The primary oral beta blocker options you’ll see include: atenolol, metoprolol, nadolol, propranolol, and sotalol. If you haven’t noticed, agents that have beta-blocking properties tend to end in -olol.

(but seriously)
(jkjk)
Source: Amazon
The reason you don’t tend to see other agents is that there’s really just not a ton of literature/data on the other oral agents but they can be used.
| Oral Beta Blocker | Dose |
| Metoprolol Tartrate | 25 – 100 mg PO BID |
| Metoprolol Succinate | 50 – 400 mg PO QD |
| Atenolol | 25 – 100 mg PO QD |
| Propranolol | 10 – 40 mg PO TID or QID |
| Nadolol | 10 – 240 mg PO QD |
| Carvedilol | 3.125 – 25 mg PO BID |
| Bisoprolol | 2.5 – 10 mg PO QD |
Chronically, we give these beta blockers orally to our patients. But in the acute setting – let’s say a patient that presents in Afib with RVR – we have IV options that we can give our patients that can work quickly. IV beta blockers include IV esmolol, metoprolol, and propranolol.
Check out the below:
| IV Agent | Dose |
| Metoprolol tartrate | 2.5 – 5 mg IV bolus given over 2 minutes, up to 3 doses |
| Esmolol | 500 mcg/kg IV bolus over 1 minute; then 50-300 mcg/kg/min IV |
| Propranolol | 1 mg IV over 1 minute; up to 3 doses at 2 minute intervals |
So when might I want to use a beta blocker first line?
Using a beta blocker as a first line agent for rate control might be a great option for a patient with chronic systolic heart failure, since we know that drugs like metoprolol succinate, carvedilol, and bisoprolol actually reduce mortality in these patients. In patients like these, a beta blocker would be like “hitting two birds with one stone” – you can help rate control their afib AND treat their chronic heart failure.

When might we want to avoid a beta blocker?
Keep in mind that even though some beta blockers are beta 1 selective, they aren’t beta 1 specific. In other words, even our selective beta blockers might start hitting off-target receptors like beta-2 receptors. Beta-2 receptors are located in the lungs and when blocked by beta blockers, can cause bronchoconstriction. These agents may want to be avoided in patients with pulmonary issues such as COPD.
Usually “beta 1 specific beta blockers” can be a great option over other agents – like our non-DHP calcium channel blockers – in our patients with “soft” (aka on the lowish end) blood pressure, however, our non-selective agents still have the ability to antagonize the alpha receptor in the periphery, causing vasodilation and hypotension. And keep in mind even our “beta specific” agents lose their specificity at higher doses, and also have the propensity for decreasing blood pressure.
Non-Dihydropyridine Calcium Channel Blockers
Next we move on to our non-DHP calcium channel blockers – verapamil and diltiazem.
We do NOT use DHP calcium channel blockers for rate control – only non-DHPs.
Just like our beta blockers, our non-DHP CCBs are available both orally and intravenously, so can be used for both chronic (oral) and acute (IV) treatment.
Like our DHP calcium channel blockers, our non-DHP calcium channel blockers block calcium channels. However, our non-DHPs are unique in that they also work directly on the AV node to slow heart rate. Which is totally the reason why we use them for rate control.

When might I want to use a non-DHP calcium channel firstline?
Non-DHPs are a great option for patients who would not be good candidates for beta blocker therapy and also for patients with concomitant hypertension since our calcium channel blockers tend to be a lot more potent than our beta blockers at decreasing hypertension. This is another example of our “two birds, one stone” vibe.
When might we want to avoid a non-DHP calcium channel blocker?
Non-DHPs would not be a good option (and should not be used) in patients with systolic heart failure and decompensated HF because of their negative inotropic effects. In other words, one of the effects of the non-DHPs is that they decrease the force of contraction within the heart. In patients with systolic heart failure (aka HFrEF) who already have such a shitty squeeze, the last thing we want to give them is an agent that will weaken that contraction even more.
They can be used in patients with HF with preserved ejection fraction, however, since these patients have no issue with contraction power.

Other Options Potpourri: Digoxin, Amiodarone, Dronedarone
So, beta blockers and our non-DHPs really represent our two first line options for rate control. But there are also other options that can technically be employed for rate control.
Digoxin
Digoxin can also be employed for rate control in Afib, though it is not one of our first line agents. Even though IV digoxin can slow ventricular response, it takes >1 hour and its effect does not peak until ~6 hours after administration. Because of this. it is not a great agent to use when rapid rate control is wanted.
During chronic oral therapy, digoxin can slow the resting heart rate, but it is not effective at controlling the ventricular response during exercise. But worry not, other agents, such as non-DHP CCB or beta blockers can be used in combination with digoxin to improve rate control during exercise.
So, when should I consider digoxin?
Digoxin may be an attractive agent for chronic rate control in patients with systolic heart failure, as it is one of the few rate control agents that does not have negative inotropic effects (it actually increases inotropy). It is also fairly blood pressure neutral so for our patients with softer blood pressure, it can be a decent option to use.
What else should I know about digoxin?
Digoxin is pretty cool – I could honestly do a whole blog post on it. It originates from the foxglove plant which they literally sell all the time at Home Depot. Like I literally saw it last weekend.

The big thing to know about digoxin is that it is what we call a narrow therapeutic index drug. Which means it has a very narrow range (in terms of blood concentration) where it is therapeutic. Give too much, and you become supratherapeutic – and can get bad side effects; too little drug and it becomes ineffective.
Digoxin is renally eliminated, so it should be at the forefront of your brain when your patient develops acute kidney injury and/or figuring out a starting dose.
Obesity also doesn’t matter when dosing digoxin – digoxin doesn’t distribute into fat, but rather into lean muscle tissue.
Digoxin can be used for both afib and systolic heart failure. The rule of thumb is that you never give a load a patient for systolic heart failure – IV loads are only for afib.
Our goal concentrations differ based on disease state – patients that have systolic heart failure get a goal trough of 0.5-0.9 ng/mL; patients that only have afib can go up to 1.2 ng/mL.
What if your patient has both systolic heart failure and afib?
You’ll still reach for that lower 0.5-0.9 ng/mL goal.

The other thing to think about is when to get a level.
We monitor digoxin levels based on their trough (aka their lowest concentrations) – so the ideal level would be taken immediately prior to a patient’s next scheduled dose.
The other question is when to get a level.

…you just said that. No I literally mean when. Before the second dose? sixth? twentieth?
In pharmacology, we ideally want to get a drug concentration level once the patient reaches steady state, or Css. This is when the rate in = the rate out and the concentration is finally equalized.
The rule of thumb is that it takes about five half lives to reach steady state.
If you look up the half life of digoxin, you’ll find that it ranges anywhere from 36 hours to up to 5 days in patients with renal failure. If you multiply this by 5, you’ll find that Css will take anywhere from 180 hours (7.5 days) to 25 days (!)

This should kinda make sense since this is the whole reason why there is an option for a load for digoxin – to help us reach Css faster.
My main point about digoxin is that Css takes longer than you might expect. There’s nothing wrong with getting an early level if there are concerns, but just keep in mind that it might be underestimating the patient’s level once they hit Css. In other words, don’t freak out and increase their dose suddenly if you get an early lower level.

Source: GIPHY
As I said before, digoxin toxicity is totally a thing. Biggest side effects to look out for are GI upset, bradycardia (duh), AV block, and hyperkalemia. Just an FYI we do have an antidote for digoxin toxicity but that’s a whole other talk for another day.
There is also some data showing an association between digoxin and mortality in its long term use. In the famous AFFIRM trial, digoxin was associated with an increase of mortality, irregardless of sex or HF. The DIG trial also found that serum levels >0.9 ng/mL in HF patients were associated with increased mortality as well. However, in another AFFIRM subgroup propensity-matched analysis with paroxysmal and persistent AF, there was no increase in mortality or hospitalization in those taking digoxin as baseline initial therapy. The jury isn’t really out on digoxin yet, but as of right now, it still has a place later down in rate control treatment.
Amiodarone
Amiodarone is an antiarrhythmic drug that has both beta blocking AND calcium channel blocking properties and can depress AV nodal conduction as well. Although you CAN use it in patients for rate control, it is less effective than our CCBs and other agents and also requires a longer time to achieve rate control (e.g. 7 hours versus 3 hours for diltiazem)
Amiodarone also has a large volume of distribution which means it needs a load.

And I mean large.
What do I mean by volume of distribution? Vd, or volume of distribution, is another one of our handy dandy pharmacology principles. The technically definition is that it “represents the apparent volume into which the drug is distributed to provide the same concentration as it is currently in blood plasma”.
If you have a brain like me, I have no idea what I just said.

The way I think about Vd is by thinking about how much of the drug I put into the bloodstream will stay in the bloodstream. Drugs that are hydrophilic (aka they like water) tend to have low Vds which means that if I inject “X” mg of drug into your bloodstream, it’s going to mostly stay there in your blood.
Drugs that have high Vds tend to be lipophilic (aka they like fat/tissues). This means that when I inject “X” mg of drug into your bloodstream, it’s going to instead leave the bloodstream and deposit into your tissues. The higher the Vd, the more total amount of drug I have to “load” you with until your tissues are considered “saturated” with that drug and further doses will finally want to stay in your bloodstream.
For amiodarone, patients need a total of 8-10 grams (!) of drug to finally saturate their tissues. In other words, until I reach that 8-10 gram mark, any drug I give will just deposit into the tissues. Once we get past this loading phase/dose, all further doses should in theory stay in the plasma and reach Css.
If you give a high intravenous loading dose, you may reach Css sooner but you may see worsening hemodynamics in patients with recent decompensated HF.
Long term, amiodarone also has a ton of side effects and DDIs. And I mean a ton. Pulmonary toxicity, thyroid toxicity, liver toxicity, ocular toxicity – even dermatologic toxicity (it can make your skin blue).
Because amiodarone is so lipophilic, it also has a loooooooong half life – around 30+ days. Which means that in chronic therapy even if you stop your drug – that amiodarone isn’t going anywhere anytime soon.

Dronedarone
Our last agent is dronedarone. If you notice, dronedarone sounds a lot like amiodarone. Dronedarone is essentially a modified analog of amiodarone – if we want to get more into the weeds, it is amiodarone minus its iodine moieties.
Dronedarone has been shown to be an effective rate control agent in AF and can reduce the resting HR by around 12 bpm and can improve exercise control.

However, the big things to note about dronedarone is that it should never be used in permanent AF since the PALLAS trial found that it can increase the risk of heart failure, stroke, CV death, and unplanned hospitalizations.
It should also be avoided in patients with heart failure and left ventricular systolic dysfunction because it increases the risk of stroke, MI, systemic embolism or death Increased Mortality after Dronedarone Therapy for Severe Heart Failure Trial).
Pre-excitation Syndrome: What the heck is that?
There is also something VERY IMPORTANT to know when we are talking about afib – something known as pre-excitation syndrome.
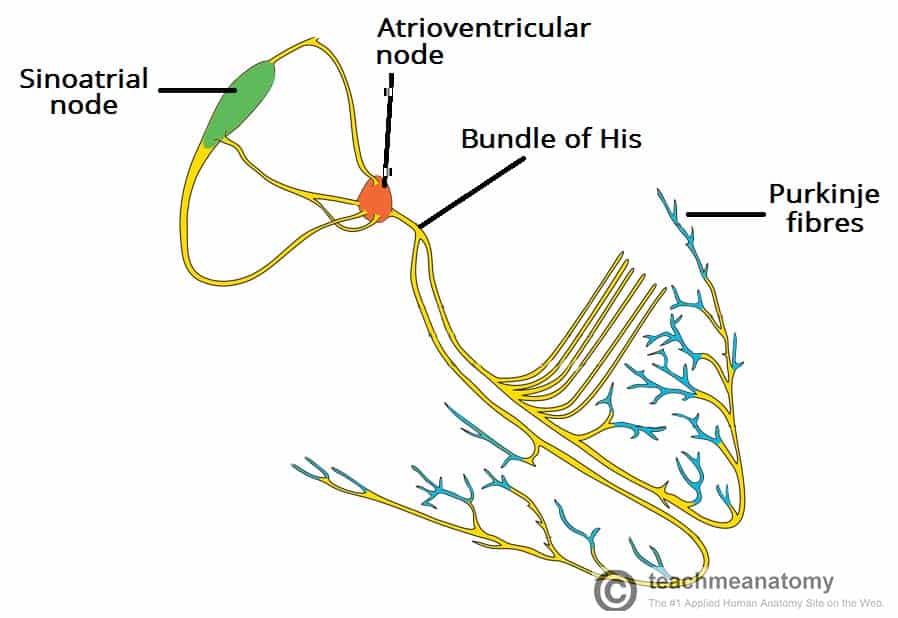
Pre-excitation syndrome is a heart condition when the ventricles are activated too early. This is caused by the presence of an abnormal electrical connection (aka accessory pathway). Like we discussed before, normally the atria are electrically isolated from the ventricles and those electrical signals must pass through the AV node.
In patients that have “pre-excitation”, they have at least one other distinct pathway that electricity can conduct through. Remember that the AV node naturally takes a pause as it conducts through the AV node. This ensures that the atria have enough time to empty their blood to the ventricles prior to the ventricles contracting so we can have good blood flow out. This abnormal accessory pathway does not have such a mechanism so those electrical stimuli immediately pass through the ventricle and will do so faster than the normal AV/bundle of His system.
In patients that have both pre-excitation and AF – all of our above options should not be used because they all act on the AV node – by depressing the AV node further, they might actually increase the ventricular response (aka electrically might preferentially go down that accessory pathway) and may result in v fib. In these patients, drugs that decrease/slow AV nodal conduction (beta blockers, calcium channel blockers, digoxin, amiodarone) should be avoided.
This is an important concept so let me say that again in another way. Normally when patients get atrial fibrillation, those ectopic electrical signals have to pass through the AV node. The AV node is an amazing gatekeeper and prevents most if not all of these electrical signals from being sent to the ventricle and triggering ventricular systole/contraction. The AV node slapping away these signals prevents patients from allowing their ventricles to contract as fast as the atria are – after all if they contracted as fast as the atria, we wouldn’t just have afib, we’d also have vfib – aka a cardiac arrest.
Patients that have pre-excitation syndrome have another route/pathway that electrical signals can choose to pass through from the atria to conduct through to reach the ventricles. This pathway is independent of the normal AV nodal pathway, and does not have the safeguards that the AV node has. So if a patient has both Afib and pre-excitation, those atrial ectopic afib signals can pass through either the normal AV nodal pathway or can travel through this extra pathway they have.
This is a problem – because of this extra pathway that doesn’t have any safeguards, these patients can present with very high heart rates since a lot of these ectopic signals may be passing through this extra pathway.
However, some of these signals are still passing through the AV node and being blocked and/or slowed.
Now, by giving a drug that depresses AV nodal conduction, we are basically turning that AV node pathway off (or at least turning it down a lot). Because of this, you have the possibility of ALL of these ectopic conductions to travel down this aberrant accessory pathway and induce – you guessed it, ventricular fibrillation aka cardiac arrest which can easily lead to death. By suppressing the AV node, we are basically telling those electrical signals to preferentially conduct through that extra pathway.
In the rare case of a patient with both pre-excitation and afib, procainamide is the pharmacological option we use.
Our Last Resort…
A last resort option includes AV nodal ablation with permanent ventricular pacing. We might opt to do this if we’ve tried and failed pharmacological therapy and rhythm control isn’t achievable. This strategy will basically render your AV node useless and not let any of those signals from the atria conduct down to the ventricles. Instead, you will have separate electrical stimulation provided to the ventricles with a pacemaker that is implanted.